



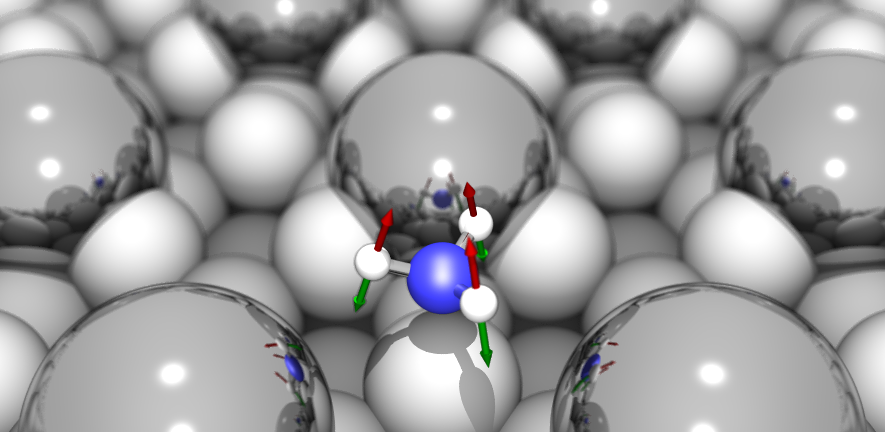
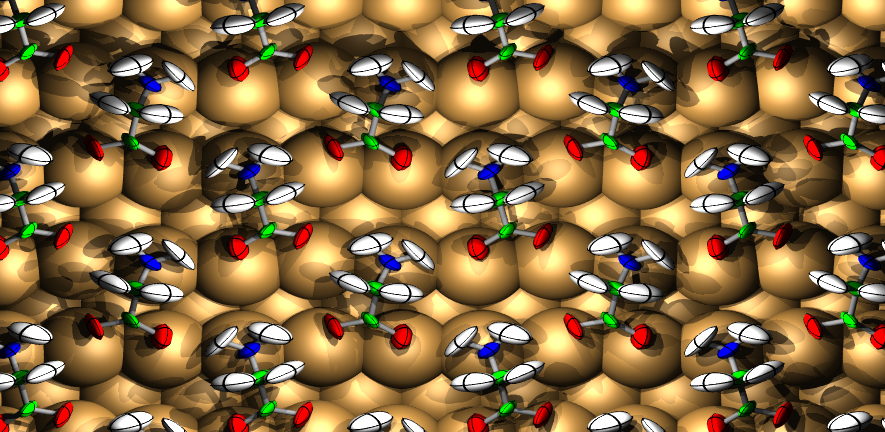
The Surface Science Group led by Dr Stephen Jenkins conducts research into fundamental properties and processes associated with crystalline surfaces. Our interests range from relatively esoteric topics such as surface symmetry, to more applied properties of direct relevance to real-world problems including catalysis, corrosion and tribology.
The group tackles its investigations through a combination of experimental and theoretical approaches. We perform experiments in ultra-high vacuum on well-defined single-crystal surfaces, using kinetic, spectroscopic, calorimetric, diffraction and scanning probe techniques. Our theoretical calculations are performed at the level of first-principles Density Functional Theory, benchmarked against experimental data. This close coupling of experiment and theory is a unique feature of the group, and allows us to obtain a far more complete understanding of surface phenomena than either would provide alone.